
AI is transforming medical imaging, but inconsistent regulations worldwide create challenges for safety, equity, and innovation. Key takeaways:
- EU: The AI Act enforces strict risk-based rules for medical AI, with penalties for non-compliance. High-risk tools face rigorous audits, but regulatory sandboxes allow limited testing before full deployment.
- USA: The FDA uses a flexible, case-by-case approach, focusing on risk classification. Predetermined Change Control Plans streamline updates for AI tools, but ethical safeguards vary.
- UK: The MHRA promotes testing through the "AI Airlock" sandbox, lowering pre-market barriers. However, the absence of a dedicated AI law leads to less regulatory clarity.
- Asia-Pacific: Policies differ widely. China enforces strict classifications, Japan provides ethical guidelines without penalties, and Singapore emphasizes cybersecurity. South Korea's Basic AI Act introduces risk-based oversight in 2026.
Key Challenges:
- Global Disparity: Only 15.2% of countries had AI-specific laws as of late 2024. The Global South lags, with 60.3% lacking formal oversight.
- Liability Issues: Questions remain about responsibility when AI errors occur, slowing clinical adoption.
- Fragmentation: Misaligned regulations increase costs and complexity for companies operating across borders.
To navigate this fragmented landscape, companies should align with international standards like ISO 13485 and prioritize region-specific compliance strategies.
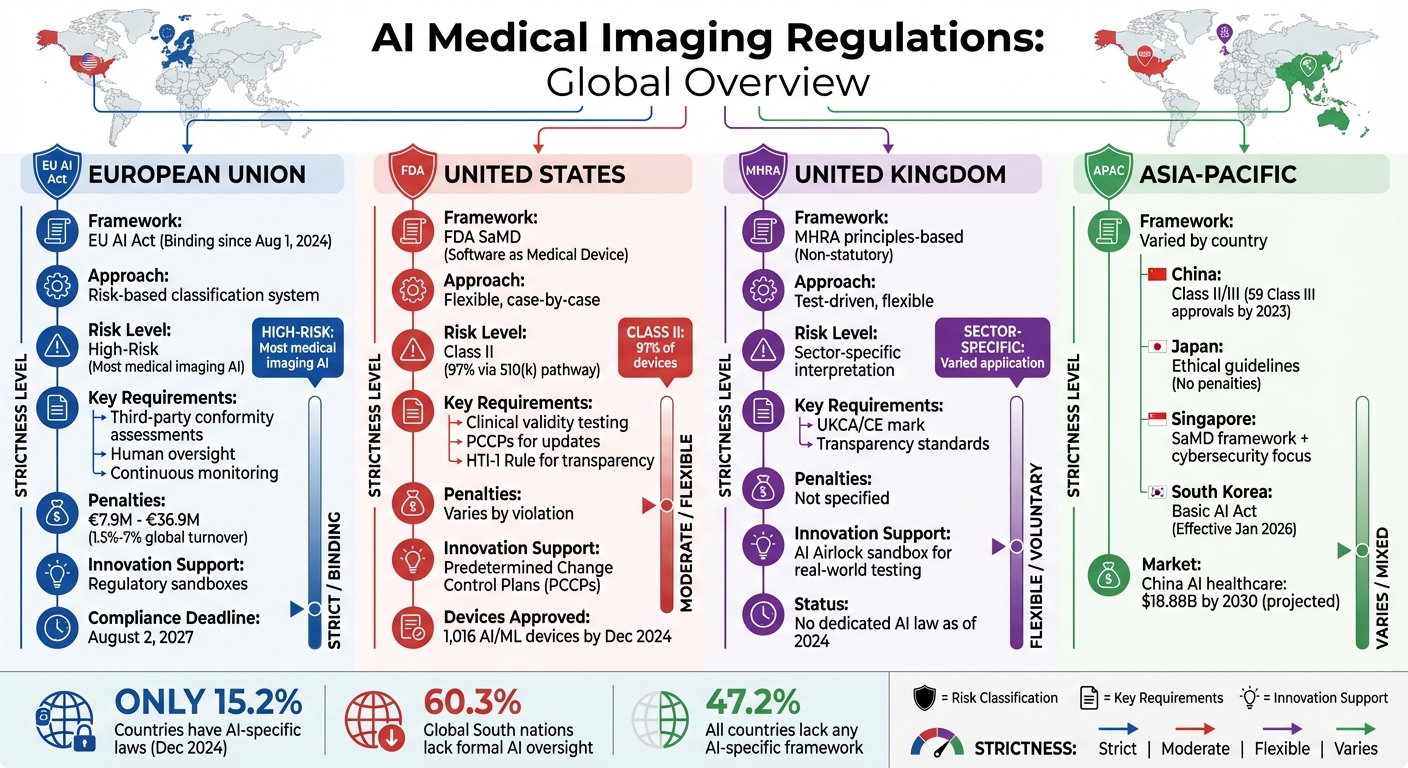
Global AI Medical Imaging Regulations Comparison: EU, USA, UK, and Asia-Pacific
AI Regulation & Transparency in Radiology with Dr. Hugh Harvey (ECR25)
sbb-itb-f5765c6
1. European Union AI Act
The EU AI Act officially became binding across all 27 member states on August 1, 2024, marking the first comprehensive framework for regulating AI in the EU. This legislation uses a risk-based classification system to categorize AI applications into four levels: Unacceptable Risk, High Risk, Limited/Transparency Risk, and Minimal Risk. When it comes to medical imaging, most AI-driven diagnostic tools automatically fall under the High-Risk category due to their classification as Class IIa or higher under the Medical Devices Regulation (MDR) 2017/745 [3][4].
Risk Classification
AI tools used in areas like diagnosis, treatment planning, or clinical decision-making are automatically considered high-risk. This categorization underscores the potential for these systems to directly affect patient safety and health outcomes. Additionally, the Act mandates that any AI system generating synthetic imaging content must include watermarks or disclosures to inform users that the content is AI-generated [7][9].
Compliance Requirements
Given their high-risk status, these systems must comply with strict regulatory standards. Developers are required to implement continuous risk management processes, maintain thorough technical documentation, and design systems that are interpretable to meet audit requirements before deployment in European hospitals [3]. Human oversight is also a critical component - systems must allow for human intervention when needed [3][4]. Furthermore, third-party conformity assessments are mandatory to verify both performance and safety prior to market entry, and post-market monitoring must track the system's clinical performance in real-world settings [3].
"High-risk AI systems, including medical devices under the EU Medical Device Regulation (MDR), must now meet new standards regarding fairness, bias control, equity, transparency, and training data." - medRxiv [4]
Non-compliance comes with hefty penalties. Fines range from $7.9 million (or 1.5% of global turnover) to $36.9 million (or 7% of global turnover), depending on the nature of the violation [11]. These penalties apply regardless of where a company is headquartered, highlighting the Act's extraterritorial enforcement [4].
Innovation Impact
While the Act imposes strict compliance requirements, it also includes measures to encourage innovation. One key provision is the establishment of regulatory sandboxes at the national level. These controlled environments allow companies - both startups and established players - to test imaging AI in real-world clinical scenarios before a full-scale launch [3][9]. However, the administrative workload has led some international developers to delay launching their products in the EU due to the complexity of compliance [9][10].
Timelines
The Act's rollout follows a phased schedule to give the industry time to adapt. Prohibitions on unacceptable AI practices were enforced starting February 2, 2025, while rules for general-purpose AI and governance structures came into effect on August 2, 2025. For high-risk medical imaging systems, the critical compliance deadline is set for August 2, 2027 [4][9]. However, discussions around the "Digital Omnibus" proposal may extend this deadline to December 2027, offering developers additional time to meet the requirements [9].
2. United States FDA Framework
Unlike the European Union's structured approach, the United States takes a more flexible, case-by-case approach to regulating AI in medical imaging. The FDA oversees AI-driven medical imaging tools as Software as a Medical Device (SaMD) under the Federal Food, Drug, and Cosmetic Act. By December 2024, the FDA had approved 1,016 AI/ML-enabled medical devices, with radiology leading the way in AI/ML clearances [4][13]. This framework emphasizes a risk-based strategy, aiming to balance patient safety with the rapid pace of technological change.
Risk Classification
The FDA uses a risk-based classification system to determine the level of oversight required for AI/ML imaging devices. Devices are grouped into three categories: Classes I, II, and III. Most radiology AI/ML devices fall under Class II (Moderate Risk), which requires "special controls" tailored to each device to ensure safety and effectiveness [13]. Class III (High Risk) devices, on the other hand, are those that could result in serious harm or death, requiring the rigorous Premarket Approval (PMA) process [13].
Around 97% of devices are cleared through the 510(k) pathway, which involves demonstrating "substantial equivalence" to an existing device. For novel devices without an existing predicate, the De Novo pathway is used. For example, in February 2018, Viz.ai received De Novo clearance for its acute stroke large vessel occlusion (LVO) detection device. This established regulation number 892.2080 and the QAS product code. By July 2022, 30 other devices had used the Viz.ai device as their predicate for 510(k) clearance [13]. These classifications guide the compliance measures that developers must follow.
Compliance Requirements
Developers of AI/ML devices must undergo rigorous testing to demonstrate clinical validity and technical performance [1]. The 510(k) pathway has faced criticism for sometimes mismatching the functions of AI tools with those of their predicate devices [13]. To address this, the FDA introduced Predetermined Change Control Plans (PCCPs) in 2023. These plans allow manufacturers to outline and gain approval for future algorithm updates and continuous learning modifications without needing to submit new applications for every change [13][4].
The HTI-1 Rule, effective February 2024, establishes requirements for AI-based decision support tools within electronic health records, focusing on algorithm transparency [12][4]. Additionally, manufacturers must engage in post-market surveillance to monitor device malfunctions and performance in real-world settings, supported by initiatives like the FDA's Sentinel program [13][6].
Innovation Impact
The FDA's lifecycle approach marks a shift from approving static algorithms to frameworks that accommodate continuous learning and real-world performance [13][4]. Between July 2021 and July 2022, the FDA cleared 126 radiology AI/ML devices, showcasing the framework's adaptability [13]. In June 2024, new guidance emphasized "Transparency for Machine Learning-Enabled Medical Devices", requiring clear communication about the AI system's logic, development, and intended use [4].
The FDA is also collaborating with Health Canada and the UK's MHRA on "Good Machine Learning Practice" (GMLP) principles to establish aligned global standards [4]. This lifecycle approach not only supports technological progress but also influences the varied review timelines for FDA approvals.
Timelines
Review timelines for AI/ML devices reflect the FDA's flexible regulatory approach. The time required depends on the complexity of the device and the regulatory pathway. For example, 510(k) reviews typically take around 90 days, while De Novo submissions can exceed 150 days due to the novelty of the devices. The PCCP framework, introduced in 2023, continues to evolve as manufacturers incorporate pre-approved plans for algorithm updates.
In October 2023, the Biden administration issued an Executive Order on AI, directing the Department of Health and Human Services to integrate the "Blueprint for the AI Bill of Rights" into healthcare regulations. This highlights the ongoing evolution of AI policy in the United States [4].
3. United Kingdom MHRA Sandbox
The United Kingdom has carved out a distinct path in AI imaging regulation, favoring a flexible and test-driven approach. Unlike the European Union's more rigid framework, the UK operates under a principles-based, non-statutory structure. It relies on sector-specific regulators like the Medicines and Healthcare products Regulatory Agency (MHRA) to interpret and apply five core AI principles within their respective domains [4]. A key feature of this system is the "AI Airlock" sandbox, which allows AI technologies to be tested in real-world clinical environments under the supervision of the NHS [15].
Innovation Impact
The AI Airlock creates a unique space where developers can test their technologies while receiving ongoing feedback from the MHRA during clinical deployment. This setup enables market access with limited pre-market data, provided the technology addresses critical clinical gaps [8]. The emphasis here is on gathering real-world evidence after market entry rather than requiring exhaustive validation beforehand.
"The sandbox is iterative and pro‑innovation, unlike the EU's binding law." - deepc.ai [15]
In July 2024, the UK government reaffirmed its commitment to AI regulation, though no new legislation had been introduced by the end of 2024 [4]. The Change Programme continues to guide the development of standards around transparency, risk management, and lifecycle validation.
Compliance Requirements
To enter the UK market, vendors must obtain a UKCA (or valid CE) mark under the UK Medical Device Regulations and work with the MHRA to meet evolving standards for transparency and bias [15][4]. Developers should also prepare for guidance on post-market surveillance and lifecycle validation, which underscores the UK's adaptive approach to regulation. This flexibility sets the UK apart in the global regulatory landscape.
4. Asia-Pacific Emerging Policies
Unlike the centralized frameworks seen in the EU, USA, and UK, the Asia-Pacific region showcases a mix of evolving approaches to regulating AI imaging. Each country has crafted its own path, from China's detailed rules to Japan's more adaptable guidelines.
Risk Classification
China has implemented one of the most structured systems. In July 2021, the National Medical Products Administration (NMPA) issued the "Guidelines for the Classification and Definition of AI Medical Software Products", which clearly outline risk levels for AI diagnostic tools. High-risk or low-maturity algorithms are classified as Class III, while others are categorized as Class II [17]. By 2023, the number of NMPA-approved Class III devices jumped from nine to 59 [18].
Japan, on the other hand, opts for a more adaptable approach. The Ministry of Economy, Trade and Industry (METI) has acknowledged the complexities of defining AI, calling it "an abstract concept" [1]. Instead of strict laws, Japan introduced the "AI Guidelines for Business Version 1.0" in April 2024. This framework offers ethical recommendations without imposing legal penalties [4]. Singapore incorporates AI imaging into its Software as a Medical Device (SaMD) framework, overseen by the Health Sciences Authority (HSA), with a strong focus on cybersecurity and system updates [16]. Meanwhile, South Korea has enacted the Basic AI Act, effective January 22, 2026, which categorizes AI tools by risk level and applies to any AI-related activity within its market [16].
Compliance Requirements
Compliance standards vary widely across the region. In China, nearly all AI diagnostic tools fall under Class III, requiring extensive technical reviews and standardized registrations. This differs sharply from the U.S., where 96% of FDA-approved AI devices are categorized as Class II [18].
"Proprietary AI algorithms pose fundamental challenges to clinical adoption... Healthcare providers express concerns about 'black box' decision-making systems that lack interpretable outputs."
– Zihuan Wang, National Engineering Research Center of Ophthalmology and Optometry [18]
Singapore's HSA mandates strict design controls and testing for connected medical devices to ensure cybersecurity. In India, the Indian Council of Medical Research (ICMR) has created ethical principles emphasizing model training checklists and clinical validation frameworks like SPIRIT-AI and CONSORT-AI [1]. Across the region, companies face hurdles such as opaque algorithms, fragmented data systems, and the lack of unified liability frameworks [17][18].
Innovation Impact
Japan's "soft law" approach, combined with initiatives like "data economic zones", aims to transform healthcare data into formats suitable for AI without imposing heavy regulations [4]. This allows startups to experiment with tools at lower compliance costs. In contrast, China's detailed regulatory system - though more demanding - provides clear steps for entering the market. For example, in early 2025, Zhongshan Hospital partnered with Huawei and iFlytek to develop a multi-modal smart hospital integrating AI imaging with clinical decision-making tools [17]. Similarly, the Second Affiliated Hospital of Zhejiang University worked with Alibaba Health to implement AI in triage and patient management systems [17].
China's AI healthcare market is expected to hit $18.88 billion by 2030, growing at an annual rate of 42.5% [18]. Chinese companies are also shifting toward subscription-based "AI-as-a-Service" models, with hospitals paying an average of $30,000 annually for installations [18]. As of 2023, Shukun Technology has secured 9 NMPA approvals for its AI imaging tools, achieving competitive standing with global players [18]. However, U.S. FDA approval typically precedes Chinese NMPA approval by 8–14 months for the same device, reflecting differences in regulatory processes [18].
Timelines
China has introduced the "2025 Measures for the Administration of High-end Medical Devices", which outlines conditional approval pathways for innovative AI projects [18]. A draft Medical Device Administration Law, released in August 2024 for public feedback, aims to regulate the entire lifecycle of AI devices, from development to clinical use [4]. Globally, as of December 2024, only 15.2% of countries have enacted legally binding AI-specific legislation, while 47.2% lack any AI-specific framework [2][4]. The Asia-Pacific region reflects this trend, with most nations still in the early phases of regulatory development.
Pros and Cons
Here's a breakdown of the benefits and challenges tied to different regulatory approaches across regions, based on our analysis.
The EU AI Act stands out for its strong ethical standards and its ability to enforce compliance beyond its borders. Any company aiming to operate in the European market must adhere to these rules, no matter where they're based. That said, developers face a significant hurdle: they must meet both the AI Act and the Medical Device Regulation (MDR) requirements, despite ongoing efforts to harmonize these frameworks [3][4].
In the United States, the FDA framework allows for agile updates through Predetermined Change Control Plans. However, it falls short in offering consistent ethical safeguards. Instead of a dedicated AI law, it relies on non-statutory executive orders, leaving gaps in ethical oversight [4].
The UK MHRA provides a more flexible system with lower compliance costs, which is particularly appealing to startups. But this flexibility comes at a price - there's less legal clarity compared to the EU, as the UK lacks a specific AI statute [4].
In the Asia-Pacific region, regulatory approaches are highly varied. For instance, China has a detailed registration system, Japan emphasizes ethical guidelines, and Singapore focuses on cybersecurity. While this diversity reflects tailored priorities, it also creates a fragmented oversight landscape, making global deployment of AI tools more complex [4][5].
A global challenge remains: misalignment between regions. This issue is especially stark in the Global South, where regulatory frameworks lag behind those in the Global North [4]. For companies deploying AI imaging tools internationally, navigating inconsistent definitions of AI, varying risk classifications, and differing liability rules increases costs and slows innovation. This fragmented regulatory environment highlights the ongoing tension between ensuring patient safety and driving technological progress.
Conclusion
While regional frameworks for AI regulation vary, certain challenges seem to crop up everywhere. Despite the patchwork of rules, some universal themes emerge: transparency, risk management, data quality, and privacy protection are priorities across the board [1]. However, the real hurdle is putting these principles into practice. As of December 2024, only 15.2% of countries have implemented legally binding AI-specific laws, leaving developers to navigate a confusing mix of sector-specific guidelines and general data regulations [4].
One of the thorniest issues is liability. For instance, if a radiologist uses an FDA-cleared or CE-marked AI tool that fails to detect an abnormality, who is responsible? This question remains unanswered globally [5]. The lack of clarity makes physicians hesitant to trust AI, fearing they might be blamed for errors caused by algorithms they don’t fully understand or control. This is particularly critical for real-time health risk alerts where immediate action is required. This reluctance slows down clinical adoption and highlights the urgent need for international regulatory collaboration.
For those working in multiple countries, waiting for global harmonization isn’t practical. Instead, there are steps you can take now to navigate fragmented regulations. Consider adopting international standards like ISO 13485 and ISO 14971 to establish a compliance foundation that works across markets [1]. In the U.S., use Predetermined Change Control Plans to update algorithms without needing constant re-approvals [14]. Meanwhile, in the EU, national regulatory sandboxes offer a way to test high-risk AI systems before their full rollout [3].
"AI-enabled products have both general requirements (like any other product) and AI-specific requirements that must be considered independently." – ITU Focus Group on AI for Health [1]
The challenges don’t stop at regional differences; there’s also a stark divide between the Global North and South. About 60.3% of Global South nations still lack dedicated AI frameworks, compared to just 18% in the Global North [4]. This disparity highlights global inequalities in AI governance. Organizations like the WHO, IMDRF, and the Council of Europe are trying to address this gap, but progress has been slow and uneven. For companies deploying AI imaging tools internationally, these disparities translate to higher costs, longer timelines, and the need for region-specific compliance strategies.
FAQs
How do AI imaging approval pathways differ across the EU, U.S., and UK?
Approval processes for AI imaging systems differ across regions, reflecting variations in regulatory frameworks.
In the United States, the FDA employs a risk-based approach. This involves premarket reviews and adherence to specific guidelines for software categorized as a medical device (SaMD).
The United Kingdom, following Brexit, continues to align with many EU standards but incorporates its own national modifications to address unique regulatory needs.
Meanwhile, the European Union focuses on risk classification under its proposed AI Act. This framework mandates conformity assessments, ensures transparency, and requires human oversight, particularly for high-risk AI applications like medical imaging.
Who is liable when an AI imaging tool makes a clinical error?
Liability for mistakes made by AI imaging tools in clinical settings depends on the specific situation and the regulations in place. Responsibility could rest with manufacturers, developers, or those deploying the tools - particularly if the AI is used improperly or altered in some way. Emerging laws, such as the proposed AI LEAD Act in the U.S. and the EU’s AI Act, are ramping up accountability. These regulations often link liability to ensuring proper use and adequate oversight of the technology.
What standards help companies comply across multiple countries?
International regulatory frameworks and harmonization efforts play a vital role in helping companies navigate global compliance. For example, the European Union has introduced AI regulations tailored to health applications, while countries like China and Singapore have implemented their own policies to address this rapidly evolving field. Additionally, organizations like the European Society of Radiology contribute by offering recommendations, such as guidelines for post-market surveillance.
These collective efforts aim to streamline compliance across borders, fostering regulatory approaches that work across different regions.